Targeted Demethylating Gene Therapy of Fibrosis
All-in-one construct dCas9-TET3CD-(target gene)-sgRNA for targeted fibrosis therapy through demethylation of the genes RASAL1, LRFN2, KLOTHO (i.e. in heart, kidney, liver, lung, cancer). Normal re-expression of these genes has been achieved successfully in vitro and in vivo as well as a reduction of fibrosis.
Challenge
The targets "hypermethylated promoter of RASAL1, LRFN2, KLOTHO" are causally involved in heart, kidney and liver fibrosis, neuronal degeneration and cancer showing reduced biological activity.
Aberrant CpG island promoter methylation and subsequent transcriptional suppression specific genes contributes causally to various pathologies. One example is RASAL1, which encodes for a Ras-GAP-like Ras-GTP inhibitor, and hypermethylation of the RASAL1 promoter leads to silencing of RASAL1 expression and increased RAS-GTP activity. The RASAL1 promoter is consistently hypermethylated in tissue fibrosis including kidney, heart and liver and also in i.e. gastrointestinal cancers. The extent of RASAL1 promoter methylation correlates with progression of kidney fibrosis in patients and mice, and rescue of RASAL1 transcription through transgenic overexpression attenuates progression of experimental fibrosis in the kidney, revealing reversal of aberrant RASAL1 methylation and rescue of RASAL1 expression as therapeutic targets to inhibit progression of kidney fibrosis. Some non-satisfactory treatment approaches based on drugs (i.e. nucleotide analogues like 5'azacytidine) or on enzymes/genes therapy do exist. While nucleotide analogues are in clinical use as demethylating therapies, they are highly unspecific and their utility is limited as second line therapy in myelodysplastic syndromes due to side effects, highlighting the need for gene specific, less toxic demethylating therapies. Members of the ten-eleven translocation (TET enzymes) family of zinc finger proteins catalyse oxidation of methylated cytosine residues (so-called hydroxymethylation), which subsequently leads to replacement of methylated cytosine residues with naked cytosine. Both hydroxymethylated and demethylated promoters result in re-expression of genes that had been silenced through CpG promoter methylation. In the kidney TET3 is the predominant TET protein. Kidney fibrosis is associated with decreased TET3 expression, and induction of endogenous TET3 expression leads to hydroxymethylation and demethylation of various genes, including RASAL1 within diseased kidneys. However, there are more than 9000 genes targeted by TET proteins within the human genome. There is a need of a gene-specific delivery of TET3 as an attractive approach to rescue expression of aberrantly methylated genes.
Our Solution
Treatment of fibrosis activating re-expression of aberrantly hypermethylated target genes RASAL1, LRFN2 and KLOTHO with gene therapy through its demethylation using a targeted nucleic acid construct. The construct is composed by a CRISPR/Cas9-based gene-specific dioxygenase by fusing a double mutated deactivated endonuclease Cas9 (dCas9) to a TET catalytic domain (TET-CD) targeted specifically to the promoter of the target genes RASAL1, LRFN2 and KLOTHO by a guiding RNA. Targeting is achieved with an inactive CRISPR system, which is a mutated Cas9 system, i.e. a dCas9 having the mutations D10A and H840A, and/or a mutated Cpf system. The dioxygenase is selected from TET protein family, i.e. TET3CD or TET1CD. Suitable vector systems are lentivirus, retrovirus, adeno virus or AAV based vector system. Successful in vivo proof-of-principle in mouse models of kidney fibrosis with the target genes RASAL1, LRFN2 and KLOTHO shows:
- target gene demethylation,
- restoration of target gene expression,
- attenuation of fibrosis.

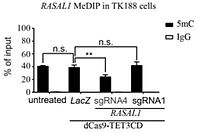
In vitro proof
The expression of the fusion protein dCas9-TET-CD guided by RASAL1 sg-RNA specifically catalyses hydroxymethylation and subsequent demethylation of the RASAL1 promoter, resulting in rescue of RASAL1 expression in fibrotic human kidney fibroblasts (TK188) in vitro. TK188 cells transduced with dCas9-TET3CD-RASAL1 using a lentivirus vector system and a control with LacZ show significantly reduced RASAL1 promoter methylation by MeDIP-qPCR assay.
Successful vivo proof-of-principle shows amelioration of kidney fibrosis in mouse mode
Experiment using a viral based delivery approach to express dCas9-TET-CD-RASAL1-sgRNA in a mouse UUO model of kidney fibrosis, which results in (i) Rasal1 hydroxymethylation, (ii) restoration of RASAL1 expression, and (iii) attenuation of fibrogenesis. 10 days after UUO mouse surgery and lentivirus injection of the dCas9-TET3CD-RASAL1-sgRNA4, the analysis have been performed. sham: stands for the un-operated counterlateral control kidney from the same mouse and is the best possible control in this system.

Rescue of RASAL1 expression. qRT-PCR results showing that RASAL1 mRNA expression was significantly induced in UUO kidney samples transduced with dCas9-TET3CD-RASAL1-sgRNA4 but not in samples transduced with control dCas9-TET3CD- LacZ-sgRNA.
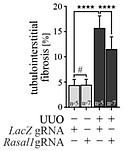 Reduction of fibrosis. Quantification of immunostained positive cells in each group, showing a significantly reduction in UUO kidney samples transduced with dCas9-TET3CD-RASAL1-sgRNA.
Reduction of fibrosis. Quantification of immunostained positive cells in each group, showing a significantly reduction in UUO kidney samples transduced with dCas9-TET3CD-RASAL1-sgRNA.
Advantages
- High specificity throught use of sgRNA.
- All-in-one construct in a a single vector, thus, increasing the efficiency.
- Rescuing expression of methylated target genes RASAL1, LRFN2 and KLOTHO.
- Use as precision medicine for disease caused by aberrant promoter methylation.
Applications
Therapy of any fibrosis, cancer or neuronal disorder due to hypermethylation of the target genes RASAL1, LRFN2 and KLOTHO. In particular useful in attenuating and treating fibrosis (kidney, heart, lung, liver..) as well as cancer (breast cancer, hodgkin lymphoma, adenocarcinoma, colon cancer, thyroid cancer, liver cancer, gastric cancer and colorectal cancer) and neuronal disorders.
Developmental Status
In vivo proof-of-principle for kidney fibrosis achieved.
Patent Status
We have a US-patent granted and a pending EP-patent application filed in the name of the University Medical Center Göttingen.
Contact
Dr. Vanessa Jensen
Patent Manager Life Sciences
E-Mail: Diese E-Mail-Adresse ist vor Spambots geschützt! Zur Anzeige muss JavaScript eingeschaltet sein!
Tel.: +49-551-30 724 149
Reference: BioT-1910-UMG
Tags: Therapie